FlashPPI2: Enhanced Model, Scaled Across 130,000+ Genomes
Since launching FlashPPI in SeqHub earlier this year (now published in PNAS), SeqHub users have predicted protein-protein interaction (PPI) networks across thousands of genomes. This has unlocked the discovery of novel interactions both within single organisms and between genomes, such as host-virus ecosystems.
Building on this momentum, we are thrilled to introduce FlashPPI2. Our latest release drives up prediction performance (AUPRC) by 17% while maintaining inference speed at minutes per genome. We've also deployed FlashPPI2 across SeqHub's database of over 130,000 microbial genomes, enabling immediate discovery of protein interactions.
FlashPPI2
The original FlashPPI framework reframed PPI prediction as a dense retrieval task enabling proteome-wide interaction prediction within minutes compared to days or months for existing methods.
FlashPPI was trained on structural complexes from the Protein Data Bank (PDB) alongside domain-domain interactions (DDIs) from the AlphaFold Database (AFDB). While powerful, experimental PDB complexes can introduce redundancy biases, and domain-level pairs don't always perfectly capture full-chain protein interactions.
Leveraging the latest AlphaFold Database expansion, which introduced 1.8 million high-confidence protein complexes, we integrated roughly 68,000 high-confidence heterodimer predictions into our training corpus. This significantly scales up the sequence and structural diversity seen by our model.
Following our rigorous validation methodology, we maintained a strict train-test split by eliminating any sequences sharing more than 30% sequence identity (at 50% coverage) with our held-out E. coli benchmark dataset.
Key Performance Gains
When tested against the highly challenging 1:100 positive-to-negative E. coli benchmark, FlashPPI2 delivers significant performance improvements over our first-generation model:
- ↑17% Improvement in AUPRC (Area Under the Precision-Recall Curve) demonstrating improved discrimination between true interactions and hard negatives.
- ↑13% Increase in Interface Contact Precision@K enabling more accurate residue-level contact prediction.
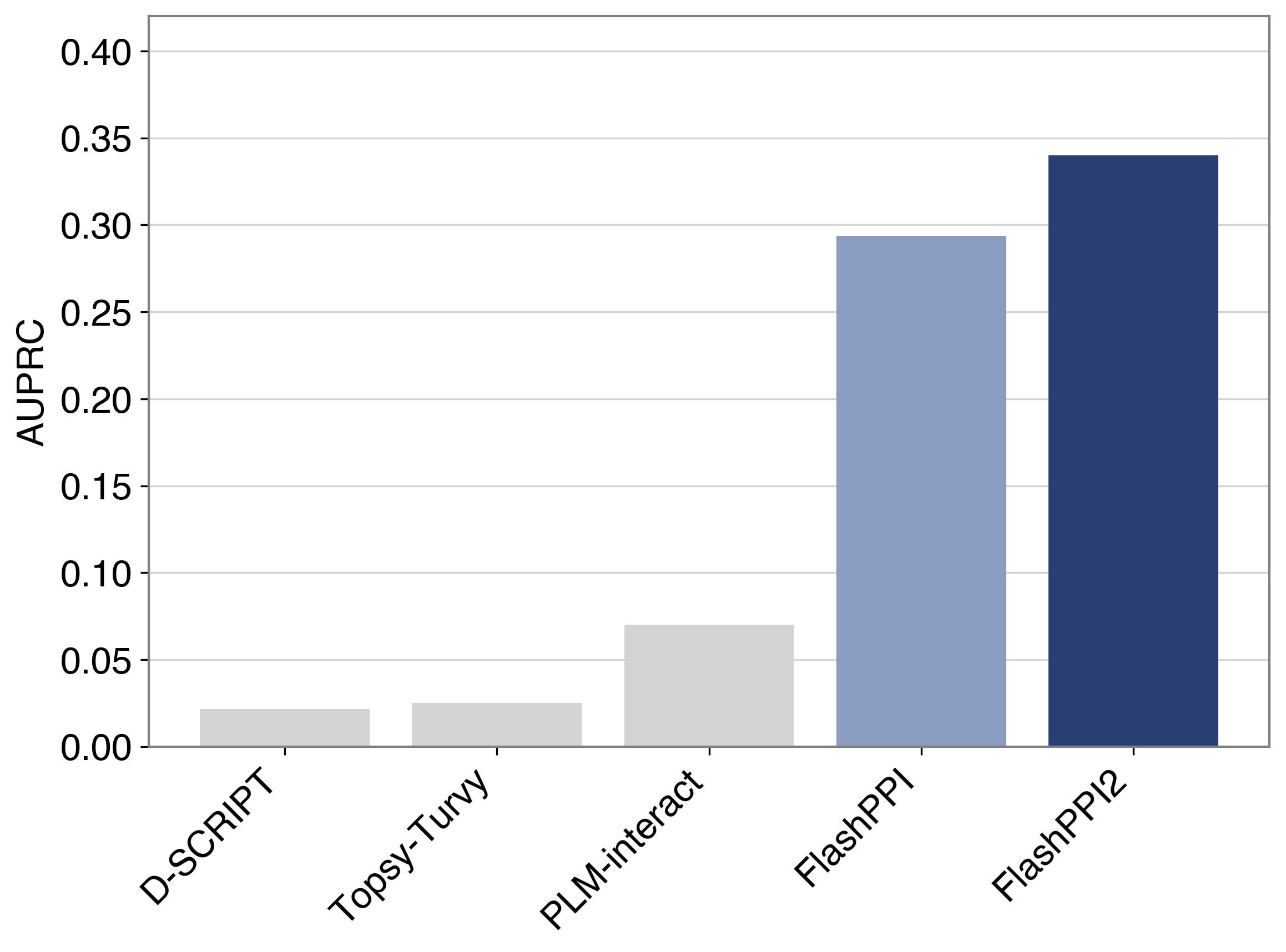
FlashPPI2 demonstrates a 17% improvement in prediction performance (AUPRC) over FlashPPI on the held out E. coli benchmark. FlashPPI2 is 3.8x more performant than the next best model PLM-interact, while enabling full proteome screening in minutes versus days.
Predicting interactions across 130,000+ microbial genomes
We've now run FlashPPI2 across the Open Genome (OG) database, SeqHub's collection of 130,000+ microbial genomes. When you run a search in SeqHub, within seconds, you can now see predicted interaction partners across this database and identify conserved interactions across genomes and species.
This scale of prediction is only possible due to FlashPPI's linear time complexity, unlike other methods requiring all-by-all comparisons. Running a comparable analysis with structure-based methods would be computationally infeasible.
SeqHub now connects your query proteins to predicted interaction partners across the breadth of microbial sequence diversity in SeqHub. This provides yet another layer of understanding of protein and pathway biological function.
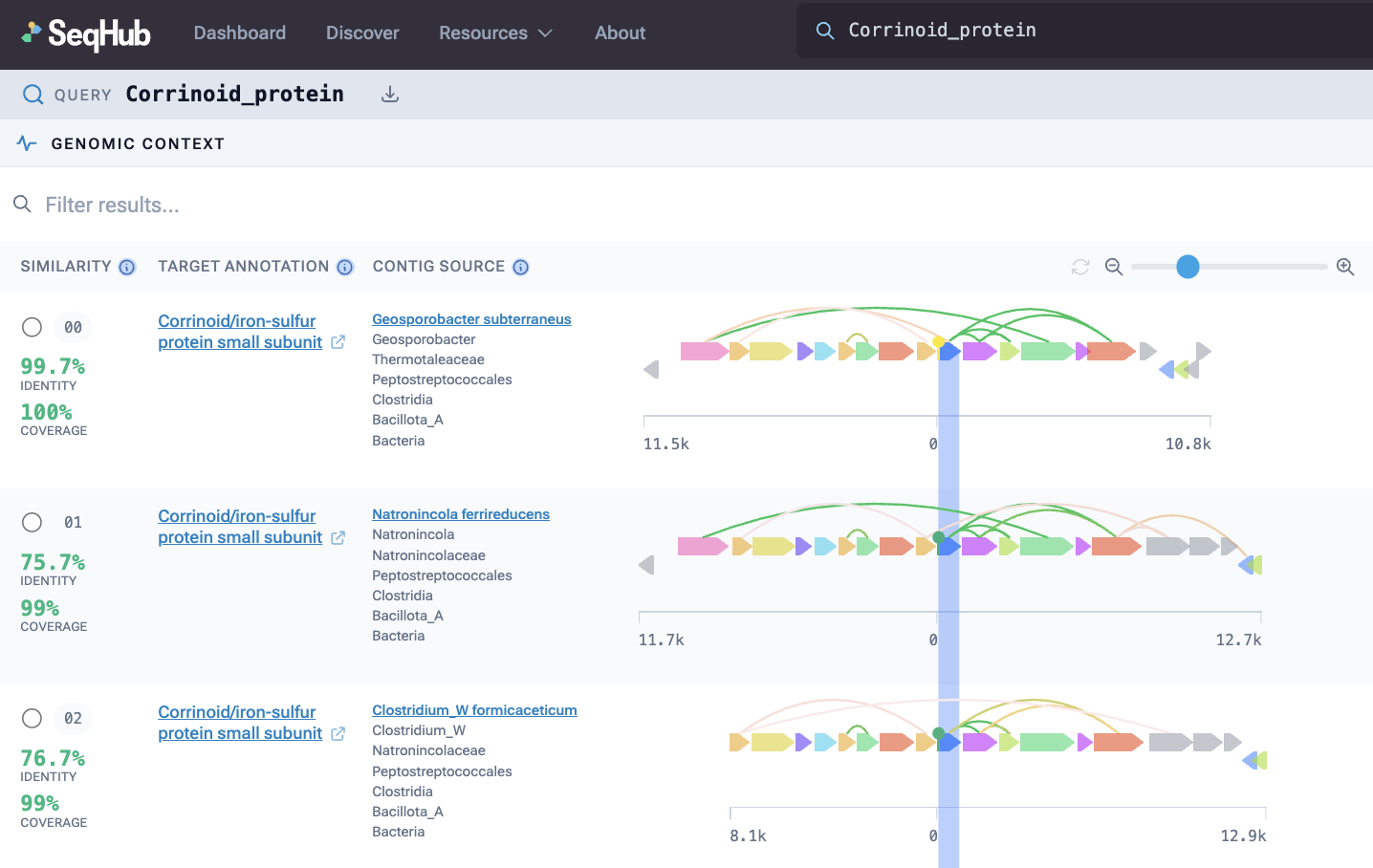
A protein search in SeqHub now shows PPIs (represented by arcs connecting genes) in genomic context across species.
FlashPPI2 is live in SeqHub
Our improved model scaled across 130,000 genomes is now live in SeqHub. With more accurate predictions, across a broader database, scientists can identify better leads when prioritizing which proteins or pathways to investigate next.
Users can run a search to find PPIs in seconds or upload a genome and predict full proteome interaction networks in minutes. If you haven't tried FlashPPI yet, you can explore results on the E. coli K-12 preview dataset to see what the output looks like before running your own.
The FlashPPI2 model is also available on HuggingFace.
Run FlashPPI2 on your protein search or full proteome in SeqHub
Get started in SeqHub →Frequently asked questions
What is FlashPPI?
FlashPPI is an open-source model for fast, proteome-wide protein-protein interaction prediction. It screens all proteins in a genome at once, in linear time, working directly from sequence rather than predicted structure. It was developed by Tatta Bio and is integrated into SeqHub.
How does FlashPPI2 compare to FlashPPI?
FlashPPI2 builds on the same dense retrieval framework as the original FlashPPI but improves training data quality and diversity by incorporating roughly 68,000 high-confidence heterodimer predictions from the latest AlphaFold Database expansion. This yields a 17% improvement in AUPRC and a 13% increase in Interface Contact Precision@K on the challenging 1:100 positive-to-negative E. coli benchmark — with no change in inference speed. FlashPPI2 is a drop-in upgrade: same linear-time proteome-wide screening, meaningfully more accurate results.
What is the Open Genome (OG) database?
The OG database is SeqHub's collection of 130,000+ microbial genomes. FlashPPI2 has now been run across the entire OG database, meaning searches return predicted interaction partners from across this collection.
How does FlashPPI compare to other PPI prediction models?
FlashPPI screens an entire microbial proteome at once in linear time, working directly from sequence rather than predicted structure. Compared to existing sequence-based PPI methods, FlashPPI delivered over four-fold performance improvement and runs 2,400x faster. Compared to leading structure-based approaches, it runs 20,000x faster. That combination of accuracy and speed is what makes proteome-wide PPI screening practical as a routine part of a research workflow.
Is FlashPPI free to use?
FlashPPI is available as part of SeqHub, which is free to use for academics and non-commercial purposes. The model weights are available for non-commercial use under a CC license; the code is Apache 2.0.
Ready to explore protein interactions at scale?
FlashPPI2 is live across 130,000+ microbial genomes in SeqHub — start discovering interactions now.