How the Rock Lab Uses SeqHub to Accelerate Discovery in Mtb and Mabs
The Laboratory of Host-Pathogen Biology at Rockefeller University studies two clinically significant mycobacteria: Mycobacterium tuberculosis (Mtb), the causative agent of TB and still the world's leading infectious disease killer, and Mycobacterium abscessus (Mabs), a non-tuberculous mycobacterium known for antibiotic tolerance and treatment failure.
The laboratory, run by Jeremy Rock, investigates how Mtb persists inside immune-competent hosts, develops antibiotic tolerance and resistance, and how related non-tuberculous mycobacteria like M. abscessus evade treatment. With roughly one third of the team on the computational side and two thirds at the bench, the lab is set up to move quickly from sequence data to experimental validation and back again.
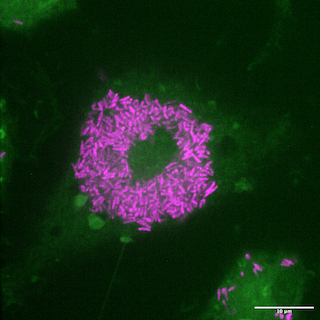
Rock Lab's image of murine macrophages (green) with Mabs bacilli (magenta).
The annotation problem in Mtb and Mabs
Much of the Mabs genome remains unannotated or poorly annotated. When a genetic screen returns a hit, it often points to a gene with no known function and no reliable annotation in any standard database. The same is true across Mtb, where large swaths of the genome lack even a hypothetical functional assignment.
Before SeqHub, the lab's workflow depended on piecing together multiple tools: UniProt for database lookup, PaperBLAST or fast.genomics for literature associations, HMMER for domain and family assignments, Foldseek for structural homology, and manual literature search to build context around homologs. Getting a useful annotation signal on an unknown protein took anywhere from 30 minutes to an hour or more, depending on how much had been published on related sequences. And even after that investment, flipping between different tools made it hard to dive deeper and get the full picture.
It wasn't very efficient to manually search what's known about homologs one at a time.
Rock Lab member
What SeqHub brings together
SeqHub brings together the signals that previously required separate tools and significantly more time — domain predictions, literature associations, genomic neighborhood visualization, structural context, and protein-protein interaction predictions — and enhances them with embedding-based search powered by gLM2, a genomic language model trained on 3.1 trillion base pairs of metagenomic data. Unlike a simple aggregator, the elements are interactive: results from one view inform and connect to others, making it easier to spot relationships that wouldn't surface from any single tool. And because the search is embedding-based rather than alignment-based, SeqHub finds functionally similar sequences even at low sequence identity, surfacing relevant biology that alignment-based tools like BLAST would miss entirely.
For a lab regularly working at the edge of what's annotated, that depth matters as much as the speed.
SeqHub has become an integral part of our workflow. As a functional genomics lab, the hits from our screens often take us in very different directions from one project to the next. Because much of the Mtb and Mabs genomes remain unannotated or poorly annotated, many of the genes that emerge from these screens are of unknown function. SeqHub is typically the first place we go to begin understanding what a gene might be doing and to identify its genomic neighbors across bacterial genomes.
Jeremy Rock, Rockefeller University
How the Rock Lab uses it
Use case 1: Functional prediction of uncharacterized genes
When the lab encounters a gene of unknown function, SeqHub is where they start building context. Embedding-based search surfaces functionally similar sequences and generates predictions; the Agent compiles and interprets that information across multiple genes at once. Literature associations surface what's known about orthologs, domain predictions and protein-protein interaction (PPI) data add further structural and functional context.
SeqHub is a great starting point to gather information about poorly annotated bacterial genomes...It's fast, easy to use, and generates outputs that are handy for downstream computational analysis.
Rock Lab member
The Rock Lab's approach is to use SeqHub for first-pass annotation and hypothesis generation, then verify with orthogonal methods where needed — a natural fit given that SeqHub is designed to complement rather than fully replace specialized tools like Foldseek or HMMER.
Use case 2: Genomic context as a functional clue
For genes in M. abscessus with no direct annotation, the genomic neighborhood view is often the most informative signal available. Seeing which genes consistently co-occur with a target across bacterial genomes can point toward function even when sequence-level evidence is sparse.
I find the 'searching for co-occurrence' feature to be very helpful. I especially like how SeqHub visualizes synteny across species.
Rock Lab member
Use case 3: Rapid PPI prediction
Protein-protein interaction prediction is computationally expensive. The Rock Lab had been running pooled AlphaFold3 to predict a set of PPIs — a process that was taking months. SeqHub's PPI feature, powered by FlashPPI, returned an initial set of predictions in minutes.
Use case 4: From prediction to the bench
In at least one case, a function predicted by SeqHub was subsequently validated at the bench. Large-scale genetic interaction mapping surfaced a gene of previously unknown function, whose interaction profile clustered with genes in a well-characterized pathway. In this case, the SeqHub functional prediction was highly aligned with the measurements from GI mapping, and was subsequently confirmed at the bench, demonstrating that the computational call translated into a real, experimentally validated function.
Annotation at the frontier
For the Rock Lab, SeqHub changed what it means to encounter a gene of unknown function. Annotation that previously required piecing together results across tools can now start in one place, with predictions that outperform what was previously available for M. abscessus. The platform gives the lab faster, deeper annotation signals on individual genes through the interface, and when the research demands something at a scale beyond what the platform currently offers, the SeqHub team has supported custom requests.
The integration also runs deeper than workflow: the lab now links directly to SeqHub from Pebble, their own gene database, reflecting how consistently the team reaches for it. The result is a lab that can move from unknown sequence to testable hypothesis faster, and with more confidence in the biology underlying that hypothesis.
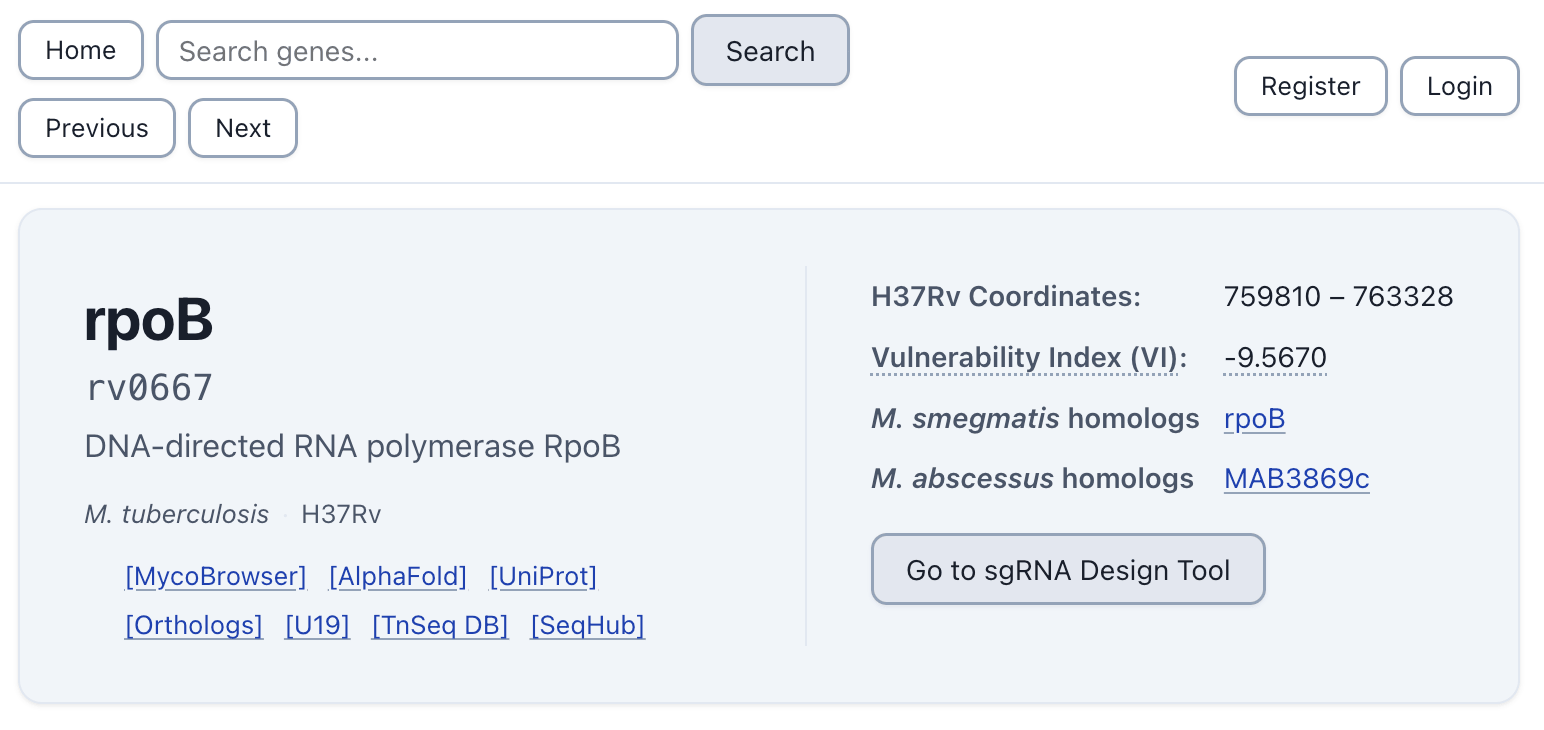
SeqHub results linked directly from the Rock Lab's database, Pebble.
Start annotating your sequences
Join research labs using SeqHub to move from unknown sequence to testable hypothesis faster.