How an NC State Microbiology Course Used SeqHub to Turn Student Sequencing Data into Published Genomes
Dr. Carlos C. Goller and TA Camila Loyola built NC State's Microbiology 360 course around a simple premise: students learn best when they're working on open questions with data they've collected themselves. The result was a bench-to-genomic analysis course in which students sequenced, assembled, annotated, and published their own Delftia genomes using SeqHub as the annotation and exploration layer.
The course
Dr. Carlos C. Goller is a Teaching Professor in the Department of Biological Sciences at North Carolina State University. His courses are built around hands-on, bench-to-bioinformatics experiences using the same tools researchers actually use.
For MB 360: Scientific Inquiry in Microbiology At the Bench, Dr. Goller collaborated with Camila Loyola, a graduate student in the Master of Microbial Biotechnology Program. Together, they built the course around a foundational premise: students learn best when they're working on open questions with data they've collected themselves.
The course centered on Delftia spp. — a genus of betaproteobacteria with a growing presence in clinical and environmental microbiology research and significant genomic dark matter still to be characterized.
The course design
Working in teams, each with their own Delftia spp. isolates, students formed hypotheses, designed their own experiments, analyzed the genome, and presented their results. Their work included extracting DNA, sequencing with Nanopore and Illumina, assembling genomes with BV-BRC, and using SeqHub to annotate and explore what they had assembled. The full pipeline looked like this:
- Practiced streaking and other core laboratory skills
- Tested growth conditions and analyzed growth curves
- Performed Biolog phenotypic analyses and co-cultured isolates
- Extracted DNA for Nanopore (long-read) and Illumina (short-read) sequencing
- Assembled genomes using BV-BRC
- Attended a live SeqHub training session before beginning annotation
- Annotated and explored assemblies with SeqHub
- Published datasets to a shared public folder on SeqHub
With their assembled genomes in hand, students explored the links between genes, pathways, and the phenotypes they had observed in the lab.
Integrating bioinformatics tools such as SeqHub and BV-BRC was part of the analysis of each group's research plan. These platforms gave students hands-on experience with real data analysis while remaining accessible enough not to become a barrier to student learning. These tools connect genome sequences back to the bacterial strains students had been working on in the lab, making the genotype-phenotype relationship something students could see and explore for themselves.
Camila Loyola, Course TA
What students were able to do
Genome annotation typically requires stringing together multiple command-line tools, a barrier in a fast-paced course where students are already learning sequencing and assembly. And, like most environmental bacteria, Delftia genomes contain proteins of unknown function that alignment-based tools struggle to resolve.
1. Annotation
Students uploaded their assembled genome FASTA files to SeqHub and received annotation results within seconds, without installing anything or writing any code. For proteins that BLAST would have returned "hypothetical" for, SeqHub's embedding-based search — powered by gLM2, a genomic language model trained on over 3 billion metagenomic proteins — found functionally similar sequences even at low sequence identity.
2. Genomic context
Because Delftia is not among the most-studied organisms, some assemblies contained genuinely novel sequence content. Students used SeqHub's genomic context view to see what was upstream and downstream of a gene of interest, whether that arrangement was conserved across other organisms, and which neighboring genes were predicted to perform what functions. They also searched for similar proteins across the broader metagenomic database, noting where their sequences appeared — in other betaproteobacteria, in environmental metagenomes, or in more distantly related taxa.
3. SeqHub Agent
To interpret results and explore gene function in context, students used the SeqHub Agent, an AI assistant that can help interpret biological sequences in context. Rather than treating annotation as an endpoint, students could ask follow-up questions about what a gene's predicted function might mean for the organism's biology.
4. Protein-protein interactions
Students also used SeqHub's PPI feature to explore potential protein-protein interactions in their assembled genomes, prompting additional questions about how genes might be working together.
We observed that students could quickly explore annotations and use the AI assistant to ask questions about their genomes. The PPI feature is a great way to explore potential interactions and helps students explore additional questions.
Dr. Carlos C. Goller, Teaching Professor, NC State
5. Collaboration and publishing
Student groups annotated their assembled genomes collaboratively using SeqHub's shared dataset features, with multiple contributors working on the same data. The final step: each student published their dataset to a shared public folder on SeqHub, depositing genomic data that other researchers can find and build on.
The published datasets are accessible at seqhub.org/ccgoller/collections/mb360spring2026
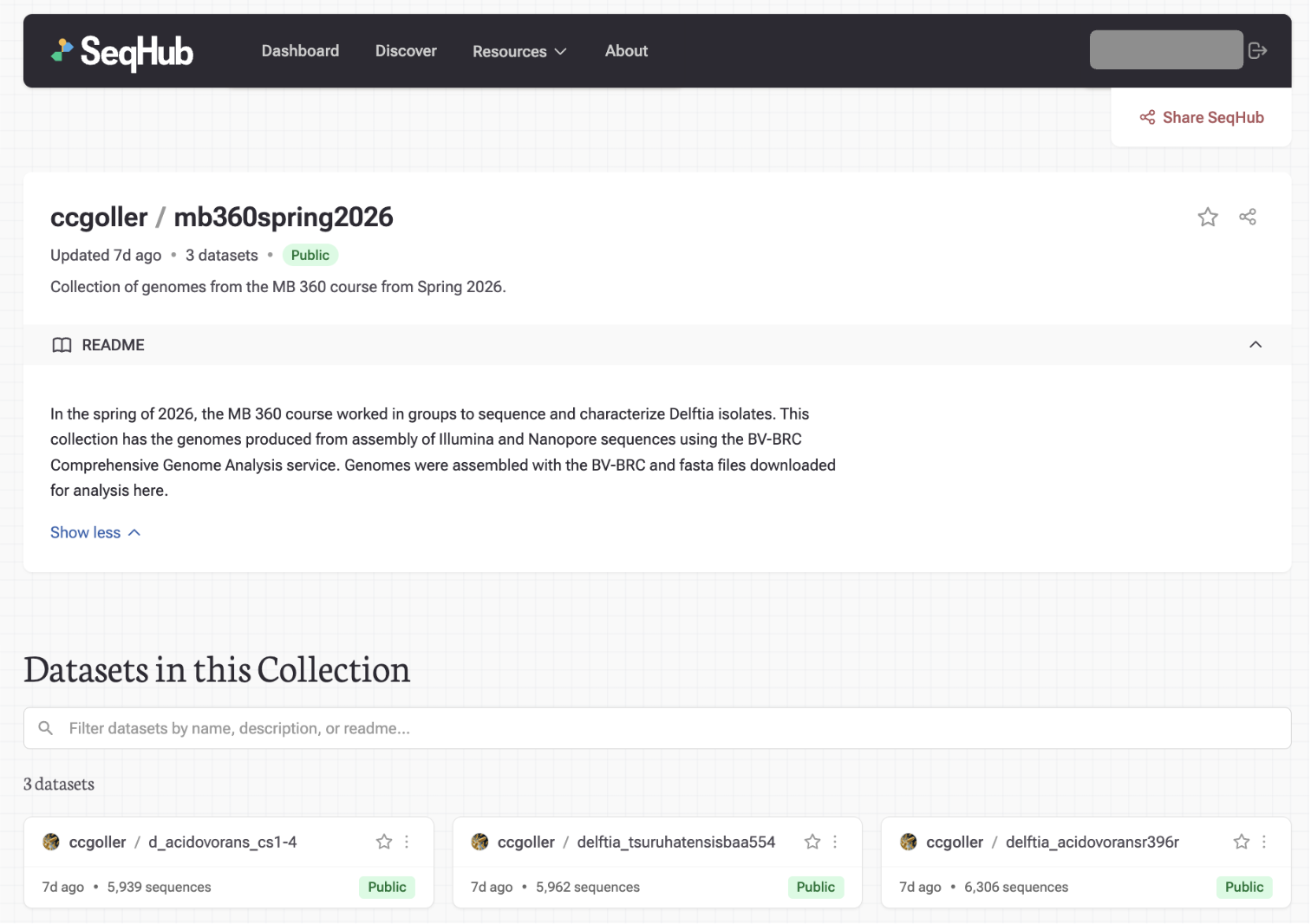
MB 360 Spring 2026 student datasets with SeqHub annotations, published to a shared public folder on SeqHub.
Using SeqHub in your own course
SeqHub is free for academic use. It accepts standard file formats for genome upload and annotation, runs in any browser without software installation, and supports publishing public datasets with read/write permissions for collaborators or students. SeqHub's documentation covers what students need to get started, and the SeqHub team is available to run a live walkthrough with your class before you begin.
Students can easily begin exploring sequences and potential pathways for their microbes. Instructors don't need detailed onboarding protocols for students…
Dr. Carlos C. Goller
Bring SeqHub into your classroom
SeqHub is free for academic use. We're happy to run a live walkthrough with your class before you begin.